Tadalafil zeigt eine konstante Resorption im Gastrointestinaltrakt, mit maximalen Plasmaspiegeln nach rund zwei Stunden. Der Wirkstoff verteilt sich gut im Gewebe und weist eine hohe Plasmaproteinbindung auf. Seine lange Halbwertszeit erlaubt eine verlängerte Wirkphase. Der Metabolismus erfolgt über das hepatische Enzymsystem CYP3A4, mit der Bildung inaktiver Metaboliten. Exkretion geschieht primär über den Stuhl. Die Häufigkeit von Nebenwirkungen steigt mit der Dosis, wobei vor allem vasodilatatorische Effekte dominieren. Ein gängiger Bezugspunkt in pharmakologischen Analysen ist cialis ohne rezept, das mit dieser Wirkstoffklasse assoziiert ist.
Consulenza dps
Laboratory of Experimental Microbiology and Epidemiology LSME, DISCAT, Institute of
Microbiology, School of Medicine, University of Genoa, Italy
Records Matching model for data survey on applied and
Salvo A. Reina , Vito M. Reina and Eugenio A. Debbia Summary Experimental microbiology provides a huge quantity of raw data which need to be evaluated and classified under a large variety of situation such marine research, environmental pollution, pharmacokinetics of antimicrobial agents and epidemiological clinical trials on infectious diseases. Almost indispensable for all kinds of disciplines is to validate, transform and correlate data clusters to demonstrate a statistically significance of results. Whether studies are for academy or industrial biotechnological purposes, the credibility of a work is strongly affected by the statistical methods and the adequacy with which those are used. Beside simple univariate analysis, many software products, either commercial or open source, are available to perform a much sophisticate statistics for discriminant and multi-factorial analysis, still the majority of scientists use partially only a minimal part of statistics methods. This is due to the high competence level requested by a multivariate approach; it is known that the choice of a test, correct distribution’s assumption, validable experimental design and not last, preliminary raw data validation are prejudicial to a good science. The need for any kind of experimentation is an analytical interpretation of descriptive evidence, and sometime the classical numerical approach is not enough because on practise applied data can not be validate or simply are incomplete. Microbiologists always wish to quickly discriminate, or correlate, groups and data clusters concerning clinical patient profiles, auditing of multi-sensor derived numbers, monitoring of a biosphere indicators on either chemical and physical parameters or dynamics of microbes population. Beside the application fields, very often the mathematical and statistical analysis is aimed to distinguish phenotypes or constraints. Basically, and practically, data are stored in spreadsheet and database files which change continuously time-by-time pending on the data collection and scopes; We here propose a Records Matching Methods (RMM) suitable for any kind of cluster analysis and pattern identification which can be use for either parametric or non parametric without necessarily state pre-process statistical assumption on variable distribution. The RMM is an application of a theoretical approach based on the Unique Factorisation Domain and is explained with an ideal generalisation model and then applied to a real-world microbiological study. Authors have used an easy mathematical formalism and discuss the possible application of the method as largely applicable to a plethora of taxonomic and phenetic investigation as well as for clinical trials and epidemiology. Prototyping of the model for a computational automated process are also described in order to realise a simple software which can infer on data by using a heuristic rules file. Keywords : Records Matching, Unique Factorisation Domain, Bioinformatics, Experimental Microbiology, Statistical Process Control, Quality Assurance, System Audit Corresponging authors : Dott. Salvo Reina, Prof. Eugenio A. Debbia (eugenio.debbia@unige.it )
Laboratory of experimental Microbiology and Epidemiology, Dept. DISCAT, School of Medicine, University of Genoa, Italy
Freelance, ICT professional, Rome, Italy
INTRODUCTION
Method here described, and its software functional specification, were thought to provide a simple tool for data calculation and experimental analysis on applied and experimental microbiology. Data analysis achieved with the method is finalised to infer, group, filter or cluster data regardless statistical assumption so that it could be applied on either diagnostics, clinics or observational measurements. Generally speaking data are a collection of records and groups of records are considered datasets. Such a scheme can be generalised to any record profile, thus a dataset is a table where rows are the samples under investigation and each column is a characteristic of the sample. The analogy of this scheme is typically a table rows by columns and each column is called “field” of the record (single row) Beside the discipline and the specific domain, dataset treated on microbiology and biotechnology needs to be analysed according several pre-process task which allow scientist to sort, classified, categorise groups of record according to descriptive criteria; afterwards, it will possible to evaluate statistics. Very often one or more datasets indicate a set of records which share common meaning and values (descriptive variable and scalar parameter respectively) and for the basic science it is essential to discriminate or associate samples according empirical criteria. A pre-process phase is indispensable especially when considering large datasets in that a validation of record integrity and coherent not null information of each filed, will impact on results credibility. Usually, dataset can be grouped and or filtered starting from a database by utilising the Standard Query Language (SQL) [10]; this compel the success of a good statistics to a high informatics competence. Moreover the SQL it is demonstrated to be effective only in evaluating “identity” criteria rather than matching groups of records according criteria such as “tolerance” and “proximity”. Applied and experimental microbiology imply several doubtfulness and “fuzzy” evidences [4,11]. Sometime the ability to approximate variables range can leverage the probability of a system adaptability (e.g. : environmental sensor automation); also, it is restrictive to use pre-determined range of significance for variables and indicators because it would be preferable to dynamically calibrate a variable or a parameter with a “weight factor” which modulate the influence and the consideration of that variable or parameter on reason of the context. Authors have already tested logics and mathematical models on several microbiological experiments concerning microorganism growth and taxonomy, Post Antibiotic Effect, MIC and genetics of quinolones [1,6,7,8,9,12,13] and these experiences drove to a unified record’s matching model. In the specific cases of marine microorganisms identification in environmental polluted mud and HIV-eukaryotic cell interaction model unsupervided Kohonen algorithms were also used [14]. Almost every experiment design used software computation. Many evolute software are available to study microbiology with multi-factorial and multivariate techniques for pattern matching such as Neural Networks, Bayesian nets and fuzzy logics. As already it was pointed up for high- performance statistic tools, artificial intelligence and reasoning software are complicate and burdensome nevertheless it would be desirable to be able to study similarities, proximity, phenotype’s varieties and cluster analysis on the every day laboratory routine. We addressed this issue by creating a simple method based on a mathematical model for cluster analysis and pattern matching; the method is practically realisable as a set of software framework which can be easily implemented by anyone regardless the programming language and the dataset file format. The method is called Records Matching Method (RMM) because it is formalised with a record profile metaphora and its example’s application is based on the recursive comparison of records which can be clustered by mean of an algorithm which use a simple template file which contains heuristic rules for each variables and parameters In order to let every one to create its software, functional specification are provided together with software documents and guide lines web coordinate. Several real world application were used by physicians for clinical trials and epidemiology applied on Assisted Reproductive Medicine ART, andrology and endometriosis surveys. In those experiences the theoretical model previously described by authors [15] was verified and tested for its simplicity and suitability so that it is now possible to provide a software front-end specification for an intuitive easy to use tool with powerful cluster analysis capability. SIMPLE MODEL FOR A SIMPLE METHOD
Either experimental or applied microbiology imply an articulated panel of factors analysis both scalars and descriptive information. Variables and parameters are generally referred to diverse typology of scales and distribution, thus record-to-record comparison as well as datasets correlation equally need parametric and non parametric statistics. We refer to a record as a pertinent set of information concerning a generic sample which is the object under investigation. Notoriously a record has a typical fields profile which is in our model globally considered as a unique factorisation index (FI); such an index has the peculiarity of being at the same time a quantitative and a qualitative expression of that specific record which can thought to as a fingerprint equivalent of the record entirely considered. If many records, hence a dataset, are serially calculated as an array of unique FI it will be possible to apply univatiate analysis on a vector of values. This simplification transform the study of complex rows by columns dataset to a series of indexes which can then evaluated according to a heuristics previously defined by the scientist empirical experience. The interaction between factorised dataset and “weighted” logics inside a heuristic file, will be the mean for which theoretical model will allow to recursively correlate FI values according a grouping criteria with dynamic and programmable range criteria. After all, the method will be represented by the ability of associating (or discriminate) samples on reason of their affinity and similarity simply because it is able to determine haw much records are diverse. We shall see that diverse could be analogously considered with the concepts of “weighted distance” of two overlapped records fingerprints (mathematical abstraction of a pattern). Because it can compare records contiguity or closeness, the model find which, and estimate how-much, a subset of records in a wider dataset table, is phenetically similar to a given record called Master Profile (MP). Generally, a MP can be a reference record which either is newly inserted in the database or is one already registered record which is assumed to be a significant paradigm. An essential step in promoting the model to a method is the definition of a heuristics logic which describe a priori the relevance of each field in the record profile. Before concepts such as correlation, association and dependence can be applied to datasets it is necessary to determine the sense and the concurrent relationships between variables relevance. In order to generalise the use of the method as much as possible we shall refer to variables and parameter with homologous fields of a record profile in that their contribution coincide with the descriptive element of our sample. On a practical base, the fields are the columns of a data table or a spread-sheet and this work will use this scheme to better explain both mathematical model and easily applicable method. Any experimental discipline uses a variety of analysis on descriptive science based on categorical information formalised in a table where rows represent records (set of studied samples) and columns which represent the characters of a sample (informative units, IU). Mathematically, a data table can be formalised ad a matrix of r by c (r x c, rows by column) and our aim is to substituting the matrix with a vector containing a series of values equivalent to each row or record. The transformation cited above it is possible with the Unique Factorisation Domain theorem [2, 3] which profit by a set of trained matrices containing the relative weights of the fields of a record so that all the range of all the possible values assumed by a field have to be classified. In fact, the matrices will be used to determine the relative distance (weighted distance) between homologous field of two records when compared and computed for their record FI. A specially useful feature of the factorisation technique is to “summarise” and “persist” a quantitative and qualitative expression of similarity in a two-records comparison by means of a delta value which sums the contribute of each single field comparison with its corresponding on the opposite record. We now introduce the definition of Matching Level or ML as the value achived each time a record-to-record comparison is complete; when operated recursively, this process originate a vector (one-dimension matrix) with all the ML values derived by the difference of two FI values. Such array of ML value will be easy to be aliquot, ranked and clusterised according to cut-off values and or arbitrary range of tolerances so that discrete bands of records can be distinguished to confine coherent groups of records on the base of phenetic closeness and relevance similarity. In the most simple case we can divide two subsets with a cut- off in the middle to separate concordant and non concordant records. This process can be repeated with arbitary cut- off to trace which samples are falling within an acceptable level of similarity
The usage of factorisation gives to the RMM a simple way of treating experimental data because the heuristic knowledge is empirically dynamically modified by the expert (heuristic Rules file, HRF) so that it can be mould and adapted to any experiment. More over, the calculation algorithm can be reiterated by systematically changing the HRF at every run and saving a corresponding results ML of FI delta’s vectors; virtuously it will be possible a supervised analysis on a well characterised samples control. This variant of the method is strictly related to the mathematical model demonstration [15] which justify an ultimate RMM usage to create calibration templates of HRF. Frequently, distinct groups of scientists share observational data typology collected with different survey, yet they wish to compare and evaluate data under a common impartial standard. Because implementation of the method is easily translatable with a software acknowledged Template of HRF can be utilised for large multi-centre audit of consensus trial, still every group could save autonomous ability of filtering, clustering and monitoring its data according specific experimental schemes. Despite its theoretical simplicity the model of RMM can lead to sophisticated reasoning software application. In short, the algorithm could indeed be ran as a self-evaluation learning system; in such a case the process is would be started without pre-defined HRF knowledge and historical repertoires could be scanned to automatically derive a set of rules automatically by inferring on raw-data regardless the stochastic and homoscedasticity assumption required by pre-process statistics. Self- referential RMM system would bring to an ideal knowledge scanning system oriented meta and cluster analysis for epidemiology. At the present time these tack can be accomplished with PCO, PCA hierarchical and cladistical generally available only in high-level statistical software packages. DISCUSSION Model Theory and applied method Mathematical treatise of the model and formal definition are disserted elsewhere [15], while in this work authors write a divulgative exposure of the theory with a minimal use of mathematical formalisms in that practical example will be oriented to experimental applied microbiology. In order to be correctly applied, the method introduced so far have to be formally defined and modelled. Before of a practical approach, we shall describe a totally theoretical example concerning the RMM. Hypothetical and imaginative example is preliminary because will simplify the comprehension and concepts realistic applicability. We premise that the field of a record have to contain non consecutive values with clearly non contiguous rank and meaning; it is also necessary to extend for each field a definition of weighted-distance which will give a direct measure of proximity or distance for a comparison of inter-fields as well inter-records entity. Let us suppose that with the notation 1) C4 = CITY = {Rome, Viterbo, Naples, Catania, Milan}(m=5)
we shall refer to the fourth field of a generic record R. The field considered is the descriptive value of the name of an Italian city [CITY] while m is the number of possible value of the field so that it will be generically noted as iM or, to be adherent to the 1) example we shall have 1a) 4M = 5, meaning with this the set of 5 possible values for the field CITY;
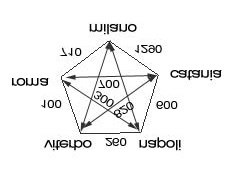
Weighted distance It is possible an intuitive to express a “weighted-distance“ between two values of the field [CITY] in terms of geographical distance expressed in kilometres; clearly, this proximity measure is reminiscent of the physical distance between cities. We now use a graph to visually represent the possible field’s reciprocal relationships. Each arc of the graph subtend a value for every couple of cities For readability, graph and its arcs are not proportional, geographical distances are intentionally approximate and original Italian names of the cities are reported; graph appears as a clear representation of the symbolic relationship of the cities with each other among those considered as possible values of the field in 1) formula. Let use the notation Gi to globally indicate the graph of all the “weighted-distances” for each couple of value of a ith field Ci in a generic record R hence we define as : 2) di (j,k) , j , for k=1., iM,
the “weighted-distance” between the jth and the kth value of the field Ci in R The graph Gi can be represent with its associate matrix defined as Mi containing the di (j,k) values for the field Ci in R: T1)
The matrix in T1) is symmetric and indeed di (j, k) = di (k, j), albeit it is possible a field typology for which the possible values d(j, k) not necessarily should have a linear correlation thus Mi would not be mirrored in its diagonal line. Each cell contains at the intersection of two possible values of the field the phenetic distance which can be interpreted as an index of affinity and similarity of two value among those possibly assumable by the field. The example described intentionally uses the geographical distance to emphasise the concept prior to apply the scheme for a more general kinds of information. The Records Matching Method (RMM) To consolidate theoretical approach we now address the model to a more specific real case. As already stressed out the practical use of the RMM is highly flexible because it can be generalised to any kind of descriptive evidences as far the scientist defines an a priori knowledgebase which, let say “informs” the algorithm on the relative significant of the information evaluated and classify all their assumable values according an indexed relevance. We now describe a case of agent-resistance experiment, still a microbiologists will immediately recognise a much large spectrum of investigation to which the RMM could result applicable with success. This case, taken as paradigm, is simple however complete since all the possible types of experimental variables and parameter, including casting variants, are treated on details. Let us recall the formalism in 1) and consider a set of fields which taken together represent a record profile. The record and its composition of fields values is obviously our sample. The RMM finality is to compare two records by determining their affinity and measuring it with a matching level. Before a record-to-record level of matching we shall explain a field-by-field matching level which is a propedeutic step; a sample is globally evaluated as result of the single contribution of each of its character whether is a variable or a parameter (field).
Consider a record R (Ci) for i=0,.7 which is a sample of an experiment concerning the estimation of the Post Antibiotic Effect (PAE) under several cultural conditions. Briefly, in vitro bacterial growth can show variable fresh outbreak after antibiotic exposure pending on cultural media and incubation time. The information characteristic collected, our fields, were registered to investigate parametric, non parametric experimental outcomes on relation to phenotype and genotypes. All indicators are also associable to a descriptive field which register the growth upshot. Schematically the record’s profile can be formalised as follows S1) C0= PAE
= {0|0.10|0.11| 0.12| 0.13| 0.14| ……………|1.0}C1= PAERange = {0-0.30 | 0.31- 0.50 | 0.51-0.60 | 0.61-0.90 | 0.91-1.2 } C2= Incubation = {60 min | 120 min | 360 min | 480 min} C3= Resistance = {R | I | S} C4= Antibiotic = {Amoxicillin | Meropenem | Ciprofloxacin | Gentamycin | Cefotaxime} C5= Fenotype = {### | PenS | PenI | PenR | EryS | EryR M | ESBL} C6= Genotype = {###| Pbp | ermB | mefA | ermTR | TEM4} C7= Growth
= {### | true | false} or {### | - | +}
Each field gives opportunity to explain all the case for which the combination of values can be translated by the model in a unique “image” which is consequence of the weighted-distance of each information therefore we describe this pharmacoresistance experiment keeping in mind that any other kind of characters can be applied as well. The first field C0(PAE), simply contains continuos values in a range of linear variability and the weighted-distance could be calculated very much as for the example previously shown in 2), thus considering a simple absolute delta between two values. On practise the distance of the two records R1 and R2 for the field C0(R1) versus C0(R2) is the algebraic difference of the values assumed by the two fields, thus if C0(R1)= 0.27 , then according to formalism in 2) d(0.45 | 0.27) = 0.18
This first example concerns linear and continuous measures and as obvious parametric variable the value itself can be appreciated as a direct measurement of geometric eucledian position. We shall soon see how the model will translate even attribute, binaries and categorical descriptive fields. The second field C1(PAERange), again, pertains to the PAE but is expressed as discrete ranks of values rather then a variable single values. For microbiologist this attitude can be reminiscent of the MIC in antimicrobial susceptibilities experiments, which indeed could be treated in the same way. The field is clearly classified according 5 ranks (restricted groups of values) so recalling the T1 matrix, we can reproduce a second matrix T2 which symbolise the theoretic graph G2 (non reported). T2) 0.31- 0.50 0.50-0.60 0.61-0.90 0.61-1.2 0.31- 0.50 0.51-0.60 0.61-0.90 2 0.91-1.2
The matrix evidentiate the relationships of the mutual combination of weighted-distance between two ranks indexes. Recalling 2) we can adapt as follows :
C1(R1)= [0.61-0.90]
C1(R2)= [0.31- 0.50]; thus d(3 | 1) = 2
In this case the delta value is calculated by using the ordinal index of the position of the rank. This is reasonable also because the ranges of the ranks, arbitrarily decide in their limits are nevertheless sorted in ascendent way. It will appears intuitive to microbiologist how limits of each rank can be arbitrarily decided pending on the experimental needs; there are no prejudices on the way the scales can be split and no forced schemes for regular length. On the contrary, diverse grouping can be decide to intentionally emphasise specific ranges.
Therefore, the phenetic distance can assume all values between 0 and 4. It can be noted how the 0 valu e means that two records R1 and R2 are identical for the field C1; moreover, this latter implication shows a first important corollary of the model which demonstrate its coherence on the contour. The field C2 allows us to consider the case of sorted and discrete variables which however do not follow a linear function. For the field C2(Incubation) expressed in minutes, the simple difference between values can be calculate in the way that the example 4) shows, still to important aspects arise. Firstly the sign of the delta value can be taken in consideration with its negative value, and in a second instance not necessarily the different relative distances from one the indexed field position could reflect the meaningful desired by the investigator on reality. Let consider these two situation starting with the ordinary notation d(120 | 480) = -360
Hence we have two possibile choices which can be adopted pending on a apriori empirical judgment of the scientist a) to use the delta value the way the are, meaning by this that the difference will be taken on absolute value 5) d(120 | 480) = -360
become |d(120 | 480) | = 360
b) to use a matrix of heuristic indexes to calculate a phenetic distance in a uniformed and predetermined way As underscored the intervals taken as absolute values between consecutive incubation time have not geometrical regularity and there are no regular proportion in the values succession; the three delta values 60|240|120, obtained the for position 60-120-360-480, are non sorted (only crescent or descendent ) and scraps are not linear. To understand how the b) situation can be favourable, we take advantage by the matrices T3a e T3b proposed as follows : T3a)
The two possibilities will be treated to gives different meaning relevance to the diverse experimental situation, still it will be processed by the RMM exactly in same way and it will be discriminative the human role. If we intend to get a linearity between incubation intervals, namely, will be all considered at the same level and we shall want only cluster and qualitative distinguish among various experiment we could use a heuristic table T3a and an example would be :
d(3 | 1) = 2
This example clearly implies proportional increments deltas and the maximal separating factor would be
[max d(0|3)] = 3 corresponding to the extremes 60 and 480 minutes.
If the microbiologist will prefer a more evident discrimination among incubation time, and even more, he wants to specifically decide which interval are more relevant to the experiments duration then an hypothetical heuristic matrix
would be th T3b. The inter-distances scheme is identical to that in T3a albeit the weighted indexes were clearly chosen according to an exponential progression. If we repeat the step 6) by applying the T3b heuristics and maintaining the same fields values we shall obtain :
d(15 | 4) = 11
It appears evident how heuristic matrices can be arbitrary rendered to fine tune the microbiologist decision which are based on the logics on his empirical experience. Detailed fields analysis of the model described so far on the first 3 fields is essentially the same for all the others hence we shall omit the formalism of the heuristic calculation to preferably exhaust all other types of information in the record structured profile designated in S1). We more briefly complete the plethora of possible fields typology and their weighted-distance casting. The field C3(Resistance) is a useful example of how attribute variables can be used as qualitative discrete inter- values. In such a case is not that relevant to conserve an ideal sorting along the three symbolic values (Resistant, Intermediate and Susceptible) therefore a simple linear heuristic matrix will adequately fit most cases in that there is no a priori preferable values direction. For the field C4(Antibiotic) are legitimate all the consideration already assured for the field C3 since values are not scalar nor oriented, still it is plausible to establish a special relevance to privilege a type of antimicrobial agent towards an other. For instance, quinolone and cephalosporin could be considered much similar and therefore much close, when compared with ampicillin. This scheme could lead the RMM resolution to a better stratification for clustering purposes because records with ampicillins will tend to segregate more centrifugally in their phenetic score. Field C5 and C6 (Genotype and Phenotype) share all previously consideration for the descriptive variables except for the peculiar value Null or symbolically [###]. It is indeed possible that either genotype or phenotype would be unknown (or not definable). This important case, again, could be a subtle clue which need to be brought on foreground to perspicuously separate samples. The C5 and C6 fields are also vital to understand a further concept of the RMM named Extended Matching Score or ExMS which makes it possible to extend the use of indexed weighted-distance by combining two concurrent fields considered to be related in some way. This is exactly the case of the genotype and the phenotype fields in that it is quite probable the expectation of having a specific genotype be associated to a phenotype. Failing this evidences should rise doubts and it would be optimal to use RMM with an appropriate logics to accurately discern. The ExMS is helpful in this case and is simple to apply because delta indexed values of two variables can simply be multiply for a factor called “enhancer” when predetermined combination of values belonging associated fields will occur. Authors have given an exhaustive treatise of the issue to include with the model the concept of fields “neighbourhoodconcurrency” [15]. At last consider field C7 which specifically issues the case of binary variables (TRUE/FALSE, YES/NO and symbolically +/-). Despite two possible values the RMM heuritic matrix would help in discriminating a third level of information’s type because the Null possibility could be indexed and samples could be diversely interpreted; possible Null values of a field could acquaint several meanings such as not measurable, unknown value or not trustworthy data. Factorial record index After a basic level of abstraction which explained the intra-record (inter-field) the model can now be scaled up to an inter-record level. The concept of weighted-distance applied to the fields relationship can be transferred to classified the entire record to step forward the RMM properly defined which will cumulate all the variable’s weights of each field of the record. This mechanism aims to substitute a sample/record with a unique number which is together a quantitative and a qualitative expression of that record. By recalling the structure in S1) we obtain a set of fields representing a record R formalised as follows : 8) R= {C0 | C1 | C3 | C3 | C4 | C5 | C6 | C7 }
eventually substituted with nominal definition
9) R= { PAE | PAERange | Incubation | Resistance | Antibiotic | Phenotype | Genotype | Growth }
We can express the content of the record R as an equivalent number called Factorial Record Index or FRI. This number has a series of features that will be useful to give a qualitative and a quantitative representation of the record. By utilising the Unique Factorisation Domain approach [2, 3] it is possible to achieve a unique number and by reversing the algorithm to go back to all the values of the field of the original record [15]. In this paper the FRI will be described with respect to the only the practical suitability with the RMM; we remind that the sum of all the weighted-indexes derived from the matricial calculation of each field of a record (e.g. : 8 and 9 formulas) is finalised to the comparison between two records. Each field inside the record profile will have a “weight”, all fields taken together, will result in a FRI. We firstly define a table called Field Weights Table or FWT which is comprehensive of 3-dimensional arrays : ordinal value of the field in R (its relative position in the record profile), its index value and its contained descriptive value. On reference of what was defined in S1) and supposing all descriptive fields as already classified in a heuristic matrix like T2), we then have a table as follows : T4) Contained
Lines dotted signify tacitly omitted fields between C4 and C6; scheme meaning remain unaltered. The fourth column is a Field’s Weight Factor or FWF and will be essential to manipulate a meticulous logics which differs importance of a field towards others. Every row of the table has a weight which act as a multiplicative factor so that the expression in 6) can be applied as
difference of two records R1 and R2 for that field; hence, that expression was d(3 | 1) = 2 for the Incubation field and now would be revised according table T4 as follows : d(3 | 1) * FWF (2|2) = 2 * 2 = 4
Basically, when a record is a case of an 2 hours incubation, its relevance during RMM is double in terms of weighted-distance with other kinds of duration. This feature of the FWF is extremely important to understand how a scientist can freely design a heuritics made with detailed rules and set up a reasoning template for the algorithmic engine of the RMM. A weighted logics, adequately prepared for a specific set of information, is a sort of optical filter which will deflect experimental dataset and re-project it on a screen as a clustered map; in a way a metaphor of the trapezoid that filter a coherent light-wave and separate in wider coloured band The case FWF (7|1) is zero, meaning by this that the Factorial Weight Index is also an effective mechanism to selectively exclude a field. This feature is useful when the investigator wants to run a RMM on a dataset considering only part of the record information; he will simply prevent the model from calculating. The seventh field C7(Growth) in T4 is a special example because can show how coherent the model would be considering other experimental situation at the edge; for instance when the detection of a value was not possible or is
not available, this does not means that there is no evidence of growth, simply the information is not available (e.g. automation and technical accidents). It is obvious the setting to zero the symbolic value of [###] will prevent the sample from being accidentally considered as [false], which rather means no growth. As last implication, the RMM ignores, namely will not compare, those records which have even only one Null FWF; only [True/False or +/-] are meaningful values. Conclusion
The proposed model of the RMM is suitable to analyse experimental dataset in the daily microbiological routine. The method is finalised to the cluster analysis and it represent a simple and customisable alternative to complex modelling software and sophisticated statistics. Its use and effectiveness is linked to the investigator that decide an a priori set of rules to determine the association level of the experimental measures studied. The rules are represented with simple and intuitive knowledge tables for each variable or parameter of a record; the heuristics can arbitrary be calibrated and adjusted so that the dataset can be scanned by the RMM algorithm which will recursively process records matching on samples’s table. Mathematical formalism of the model and its basic calculation algorithm is provided on literature [15] thus scientist who has programmatic skill, can develop its own software program by using any programming language. Virtually any type of dataset and experiments can be processed, still for practical software implementation, example of source code concerning the modelling discussed in this work is freely distributed by the authors to anyone who wish to realise the software toolkit. The auspice is that several other groups, involved in different microbiological fields, could adopt the RMM an test its efficacy. Acknowledgement Authors are grateful to the programmer Carlo Bergamini (Genoa) for the Delphi and MS-VB6 source code engineering and Franco Ameglio (Rome) for his manuscript revision of the microbiological aspects. Literature
1. Cavallero A., Reina S., Schito G.C. - Post Antibiotic Effect induced by Ofloxacin in both gram-positivi
and gram-negative bacteria. "Chemoterapia" Jul 1987.
2. FG.M. Artin, Algebra, Prentice Hall (1991) 3. D. S. Dummit, R. M. Foote, Abstract Algebra, Wiley (1999). 4. Hanai T, Honda H. Application of knowledge information processing methods to biochemical engineering,
biomedical and bioinformatics fields. Adv Biochem Eng Biotechnol. 2004;91:51-73. Review. PMID: 15453192 [PubMed - indexed for MEDLINE]
5. Pollera C.F., Ameglio F., Reina S. - Changes in serum iron levels following very high dose of cisplatin.
Cancer Chemotherapy and Pharmacology 1987
6. Reina S., Debbia E.A., Schito G.C. - Ciprofloxacin Induced Modulation of cellular grothw in activated,
normal and lymphoid established Cell Lines. The antimicrobial agent resistances: orin treatment and control.abs 70, 25 5 1991, Principato di Monaco.
7. Reina S., Debbia E., Schito G.C. - Evaluation of the post antibiotic effect induced by various antibiotics
against Staphylocossi and Enterococci. A.A.M.J. 1993
8. Reina S., Debbia E. - Genetic recombination by spheroplast fusion in Escherichia coli K12 . Cytobios by
9. Reina S., E.A. Debbia, G.C. Schito. In Vitro Cellular Growth Modulation by quinolone conditioned
medium. 93rd General Meeting, Atlanta, Georgia, USA. Session 120. Paper nu. I28.
10. Reina S., Boeri E., Lillo F., Cao Y., Varnier E.O. Automation in AIDS research and diagnostic activity: a
Local Area Network with Standard Query Language. 7th European Edition of Conference on Advanced Technology for Clinical Laboratory and Biotechnology. - ATB '91 Nov 26-11-1991 B11.
11. Reina S., Miozza F. - Knowledge Data Base System for Twins study. ACTA GENET MED ET
GEMELLOL. Ed. Mendel Institute, Rome. 1994. 43:83-88
12. Reina S., Reina V. , Giacomini M., Debbia E. - Bio-fouling and micro-organisms identification on
polluted materials: a novel Knowledge Data Base System architecture for a heuristic expert system engine. Atti congresso Internazionale dei Biologi, 22-25 settembre 1994. Vieste
13. Reina S. Il percent growth rate average (PGRA) migliora l'interpretazione dell'effetto post-antibiotico.
14. Ruggiero C., Giacomini M., REINA. S., Gaglio S. A qualitative process theory based model of the HIV-1
Virus-Cell interaction. Procedings of Medical Informatics Europe 93, Israel. ISBN 965-294-091-7 pp. 147-150.
15. Salvo A. Reina , Vito M. Reina and Eugenio A. Debbia Simple method for Records Matching for
experimental and diagnostic datasets of patient’s records. (Pre-print submitted to BioStatistics, COBRA electronic publishing network)
Editorial Subjects—In This Issue and in Previous Issues affirms our intuition that (a) patients can discern among specificmeals or rosiglitazone (Avandia) 4 mg twice daily.17 The meansensory attributes of intranasal steroid products, (b) someA1c values at baseline were 9.5% in the INH group versus 9.4%attributes have higher economic value in their avoidance for the rosiglitazone group. Af
PPCOE 2010 Program Session Code: DD-P-R DD : Date of paper presentation / poster exhibition P : Parallel block on the date R : Room for paper presentation (A, B, C) / poster exhibition (D) 08-1-A: The Agricultural Ergonomics Chair: Kwan Suk Lee , Hongik University, Korea A Korean national project of agricultural assistive technology Kwan Suk Lee, Seong Rok Chang, & Yu-Chang