Tadalafil zeigt eine konstante Resorption im Gastrointestinaltrakt, mit maximalen Plasmaspiegeln nach rund zwei Stunden. Der Wirkstoff verteilt sich gut im Gewebe und weist eine hohe Plasmaproteinbindung auf. Seine lange Halbwertszeit erlaubt eine verlängerte Wirkphase. Der Metabolismus erfolgt über das hepatische Enzymsystem CYP3A4, mit der Bildung inaktiver Metaboliten. Exkretion geschieht primär über den Stuhl. Die Häufigkeit von Nebenwirkungen steigt mit der Dosis, wobei vor allem vasodilatatorische Effekte dominieren. Ein gängiger Bezugspunkt in pharmakologischen Analysen ist cialis ohne rezept, das mit dieser Wirkstoffklasse assoziiert ist.
Bioinf.nuim.ie
JOURNAL OF VIROLOGY, Feb. 2005, p. 1836–1841
0022-538X/05/$08.00ϩ0 doi:10.1128/JVI.79.3.1836–1841.2005Copyright 2005, American Society for Microbiology. All Rights Reserved.
Evidence for Heterogeneous Selective Pressures in the Evolution of the
env Gene in Different Human Immunodeficiency Virus
Simon A. A. Travers, Mary J. O’Connell, Grace P. McCormack, and James O. McInerney*
Biology Department, National University of Ireland, Maynooth, County Kildare, Ireland
Received 30 June 2004/Accepted 10 September 2004
Recent studies have demonstrated the emergence of human immunodeficiency virus type 1 (HIV-1) subtypes with various levels of fitness. Using heterogeneous maximum-likelihood models of adaptive evolution imple- mented in the PAML software package, with env sequences representing each HIV-1 group M subtype, we examined the various intersubtype selective pressures operating across the env gene. We found heterogeneity of evolutionary mechanisms between the different subtypes with a category of amino acid sites observed that had undergone positive selection for subtypes C, F1, and G, while these sites had undergone purifying selection in all other subtypes. Also, amino acid sites within subtypes A and K that had undergone purifying selection were observed, while these sites had undergone positive selection in all other subtypes. The presence of such sites indicates heterogeneity of selective pressures within HIV-1 group M subtype evolution that may account for the various levels of fitness of the subtypes.
It has been hypothesized that human immunodeficiency vi-
tion. Recently, more biologically realistic methods have been
rus type 1 (HIV-1) may have entered humans in three inde-
developed to allow for identification of heterogeneous selec-
pendent transmissions of simian immunodeficiency virus from
tion pressure across amino acid sites and also heterogeneity
infected chimpanzees from which the three HIV-1 M, N, and
across both sites and lineages within the phylogeny (32, 33).
O lineages arose (9). Within group M, nine phylogenetically
Previous studies (4, 28) focused on searching for positive
distinct subtypes have been proposed (subtypes A to D, F to H,
selection within the HIV-1 group M subtypes by analyzing each
J, and K), with subsubtypes being proposed for subtypes A and
subtype independently and identifying amino acid sites with a
F (18). Subtype distribution varies worldwide, with subtype B
high probability of having undergone positive selection. How-
predominating in North America and Europe (15) and subtype
ever, Drummond et al. (6), referring to work by Seo et al. (20)
C accounting for more than 55% of worldwide infections (7)
as an example, suggested that positive selection seems to be a
due mainly to its prevalence in Southern and Eastern Africa (1,
minor contributor to the overall molecular evolution of HIV-1
2, 14, 17, 26) and India (21) and its increasing prevalence in
and that negative (purifying) selection imposed by functional
Brazil (22) and China (19). Biological differences including low
constraints in HIV-1 is more important than positive selection.
CXCR4 coreceptor usage in subtype C (15), decreased pro-
Here, we present an analysis of the likely selective pressures
tease susceptibility in subtype G (5), and varying subtype re-
that have affected HIV-1 group M env sequences in their di-
activity to monoclonal antibodies (13, 25) have been observed
versification from the original group M founder virus. We have
among the subtypes. The production of broadly neutralizing or
carried out this analysis by comparing each individual subtype
subtype-specific vaccines requires an in-depth understanding
to all other group M subtypes in an attempt to identify amino
of the inter- and intrasubtype evolution.
acid sites whose evolutionary history appears to be unique in
The study of the selective pressures governing the evolution
terms of selective constraints for that subtype. The identifica-
of protein-coding DNA sequences has traditionally been car-
tion of such sites yields information as to unique subtype-
ried out by comparing dN (nonsynonymous substitutions per
specific molecular traits that may also manifest as unique bio-
nonsynonymous site) to dS (synonymous substitutions per syn-
onymous site), resulting in a dN-to-dS ratio () (see reference31 for a review). An of Ͼ1 is indicative of positive selection,
MATERIALS AND METHODS
an of 1 indicates neutral evolution, and an of Ͻ1 indicates
Alignments. All available full-length envelope gene sequences were down-
purifying (negative) selection. However, if there is strong pu-
loaded from the Los Alamos National Laboratory HIV sequence database
rifying selection operating on the majority of amino acid po-
(http://hiv-web.lanl.gov) and aligned by using MacClade (12), and neighbor-joining trees were produced for each subtype by using PAUPء (23). A subset of
sitions, averaging over an entire sequence could misleadingly
the full-length envelope gene sequences for each subtype was selected by choos-
indicate purifying selection for the entire molecule even in the
ing as diverse a range of sequences as possible within each subtype based on their
presence of a small number of sites undergoing positive selec-
spread through the subtype-specific trees (Table 1). Sequences with a largedegree of similarity contain much the same information, whereas divergent se-quences will contain more information about the intrasubtype diversity. Analignment of the representative sequences of each subtype was produced by using
* Corresponding author. Mailing address: Bioinformatic and Phar-
MacClade (12). Ambiguous regions of the alignment were removed to avoid
macogenomics Laboratory, Biology Department, NUI Maynooth, May-
possible false detection of positive selection due to alignment of nonhomologous
nooth, County Kildare, Ireland. Phone: 353-1-708 3860. Fax: 353-1-708
sites. The resulting env data set contained 40 sequences and was 764 codons in
3845. E-mail: james.o.mcinerney@may.ie.
HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
TABLE 1. Representative sequences of selected subtypes
contrasted with all other subtypes using the branch site-specific models. The
branches leading to subtypes A, B, C, D, F, G, H, J, and K were labeled in a sep-arate analysis, as were the branches leading to the A1, A2, F1, and F2 lineages.
Representative sequences (GenBank accession no.)
To ensure stable results, each model was run four times using different starting
A1 .AF004885, AF457080, AF069673, AF19327, AB098333,
values, and results from the run with the best likelihood score were taken. Detection of significant sites. Codeml uses a Bayesian approach to infer the
posterior probability that a particular codon in an alignment is in a particular
category (i.e., undergoing a specific selective pressure), and generally, codon sites
with P values of Ͼ0.95 are accepted as being significantly allocated to that class.
At times, especially with the branch site models, the likelihood ratio test may be
F1 .AF077336, AF005494, AJ249238, AY173957
significant, yet no sites allocated to a particular category will consider a P value
of Ͼ0.95 as being in that category. The significant result from the LRT indicates
G .AF061642, AF061640, AF084936, AF423760
the presence of a class of sites causing significance of the model; however, the
Bayesian approach for the identification of these sites has been suggested to be
inadequate using the branch-specific models (33). In order to identify these sites
causing LRT significance, we used a site-stripping method to remove the siteswith the highest Bayesian posterior probability, and the resulting stripped align-ment was then reanalyzed (using the same models and parameters). This process
A phylogenetic analysis of the data was done by using the maximum-likelihood
was repeated iteratively until the LRT failed. Sites removed before the LRT failed
criterion as implemented with PAUPء (23) using the GTRϩIϩG substitution
were taken to be the sites contributing to the significance of the alternate model.
model as selected by Modeltest (16). In order to assess confidence in each of theinternal nodes of the constructed phylogeny, a bootstrap resampling (1,000replicates) of the data using the neighbor-joining method based on maximum-
likelihood distances was performed with PAUPء (23). Tests for saturation ofsynonymous sites throughout the phylogenetic tree were performed by using
estimates. All subtype clades in the maximum-likelihood
tree produced from the data were strongly supported by boot-
Intersubtype evolutionary analysis. The software program Codeml from the
strapping (Fig. 1), and no saturation of synonymous sites was
PAML package (30) was used for evolutionary analysis of the data set. A number
observed within the data. For the site-specific models, all LRTs
of site-specific models of codon substitution that allow for rate heterogeneityamong sites were employed, namely model 0, model 1, model 2, model 3, model
were significant with a P of 0.0005. The biologically more
7, and model 8 (M0, M1, M2, M3, M7, and M8, respectively). The null models
realistic models detected positive selection occurring at sites in
M0, M1, and M7, with dN-to-dS ratios () limited between 0 and 1, do not allow
the data with M3 and M8 allocating 14% of sites with an of
for the existence of positively selected sites. The alternate models M2, M3, and
2.4814 and 12% of sites with an of 2.58819, respectively.
M8 allow for the detection of positive selection by enabling the estimated to begreater than 1. For each of the site-specific models, all sites in the data set under
Purifying selection was observed to have occurred in the ma-
examination are allocated to one of the constrained or estimated values usingmaximum likelihood with the proportion of sites allocated to that category beingdescribed by using P values with p0 pertaining to the proportion of sites allocatedto 0, p1 pertaining to the proportion of sites allocated to 1, and so on.
Also, branch site-specific models (model A and model B), which allow for rate
heterogeneity across sites and across the tree, were employed. Model A com-putes three values and is an extension of M1 in that it limits the first two values (0 and 1) to 0 and 1 and allows the final (2), which is estimated, avalue greater than 1. Model B is an extension of M3 in that all three values areestimated. For both of the branch site models, four proportions of sites areallocated to the data set. p0 is the proportion of sites throughout the alignmentallocated to 0 with p1 being the proportion of sites allocated to 1. p2 corre-sponds to the proportion of sites with a 0 value in the background and a 2value in the foreground, while p3 corresponds to the proportion of sites with a 1value in the background and a 2 value in the foreground. The models that allowfor all parameters to be estimated are more biologically realistic than the onesthat constrain certain parameters since the evolutionary mechanisms operatingwithin a data set are never simple. Constraining certain parameters within theanalysis provides a poor representation of the data, while allowing all parametersto be estimated from the data will be a much better and more realistic repre-sentation of the data.
The significance of the alternate models (whether the alternate model is a
significantly better representation of the data than the null model) were tested byusing a likelihood ratio test (LRT) which involves taking twice the difference ofthe log likelihood between the nested models and testing for significance usingthe 2 distribution with the degrees of freedom being the difference in thenumber of free parameters between the two models. Models compared in thisstudy using LRT were M0 and M3, M1 and M2, M7 and M8, M1 and model A,and M3 and model B (for more information on the models used, see references32 and 33).
Since the branch site models operate by allowing the user to examine the
evolutionary mechanisms occurring in a particular lineage in the tree (the fore-ground) against the other lineages (the background), they provide a uniquemethod of analysis by allowing the selective constraints operating on certainsequences to be compared to the selective constraints operating on all the other
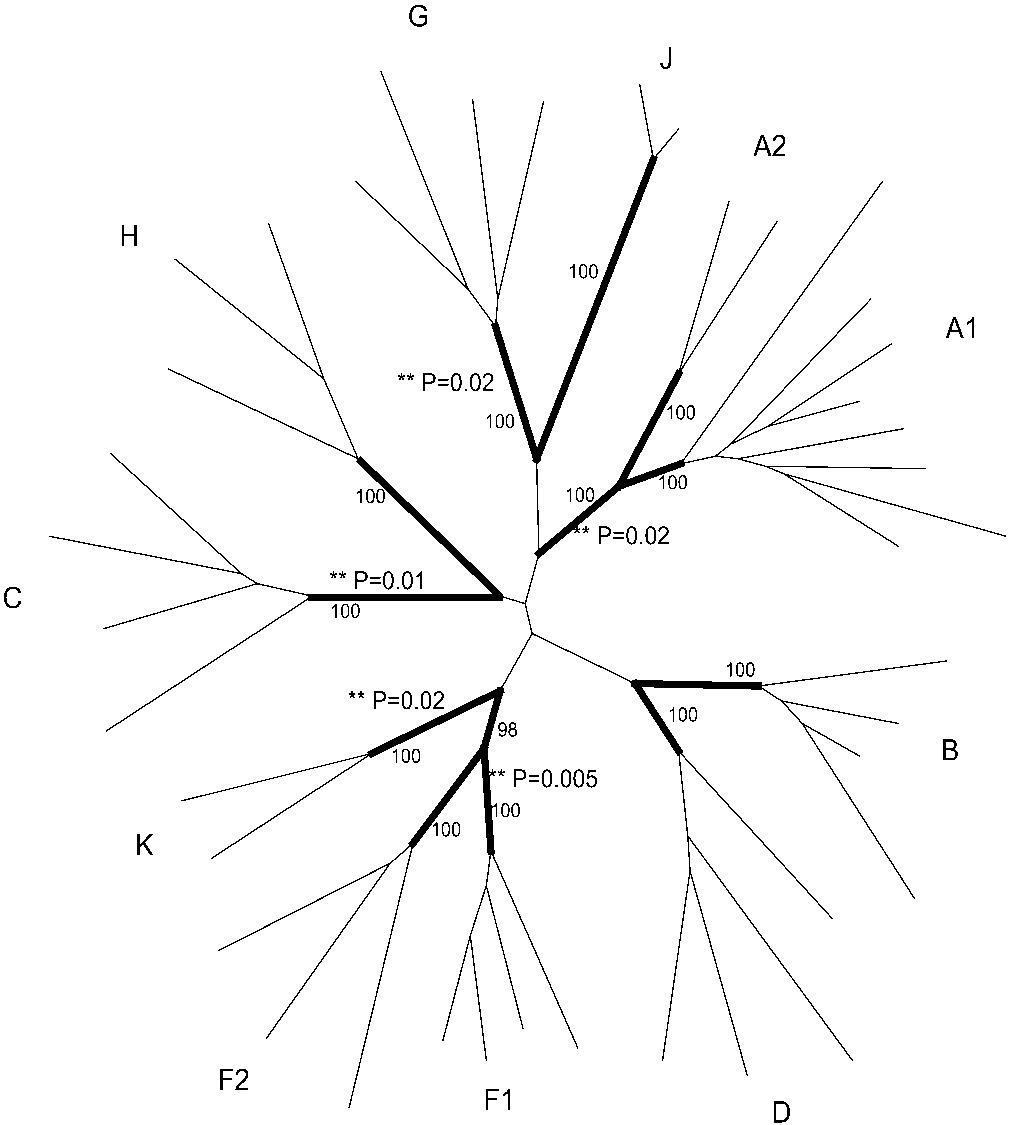
FIG. 1. Constructed phylogeny of the env data. Branches labeled in
sequences present in the data set. The site-specific models do not allow for this
boldface are the branches leading to each subtype lineage analyzed
kind of analysis, and therefore, labeling the internal node leading to each HIV-1
in this study. P values for significant branch site models are marked
group M subtype allowed comparison of the evolution of that particular subtype
(ءء P), as are the bootstrap supports for each subtype lineage.
TABLE 2. Selected sites for each subtypea
C (P ϭ 0.01), F1 (P ϭ 0.005), G (P ϭ 0.02), and K (P ϭ 0.02),suggesting that these subtypes contain a category of sites that
have evolved differently from the other subtypes. From the
branch-specific model, the subtypes’ results fell into two cate-gories. In subtypes C, F1, and G, a proportion of their sites
were observed to have undergone positive selection, whereas
all other subtypes had undergone purifying selection at thatsite (described herein as category I sites). In subtypes A and K,
a proportion of sites was observed to have undergone purifying
selection with positive selection having occurred in the other
subtypes at those sites (described herein as category II sites).
For subtypes C and F1, two sites each were allocated to cate-
gory I, while three sites were identified in this category for
subtype G. Six codons were allocated to category II for subtype
A, and 45 codons were allocated to category II for subtype K
a Category I sites were observed for subtypes C, F1, and G, while category II
The significant sites for each subtype were labeled on amino
sites were observed for subtypes A and K. Sites are described using the HXB2
acid alignments using the known protein secondary structures
b Site undergoing positive selection in at least one HIV-1 group M subtype as
for gp120 (11) and gp41 (3) (Fig. 2 and 3). gp120 structural amino acids. For the gp120 structure (Fig. c Site undergoing positive selection as determined by Yang (29). d
2), the majority of sites observed in both category I and cate-
Site undergoing positive selection as determined by Yamaguchi-Kabata and
gory II were structural sites not directly involved in known
e Site undergoing purifying selection as determined by Yamaguchi-Kabata and
gp120 functions. Within subtype K, however, a number of the
f Site undergoing positive selection as determined by Yang et al (28) for their
identified category II gp120 sites are functionally significant.
Amino acid sites 295N, 297T, and 334S correspond to a cluster
g Site undergoing positive selection as determined by Yang et al (28) in a
of nonlinear sites located on the outer domain of gp120 asso-
separate analysis of subtypes A, B, and C.
ciated with the binding of the 2G12 antibody (25). Twenty-sixresidues spanning six segments of the gp120 molecule are in-
jority of sites (86% for M3 and 88% for M8) through the env
volved in direct contact with the host cell CD4 receptor (11),
one of which (474D) was identified in category II for subtype
The branch site models were implemented to detect any
K. Sites 305K, 306R, and 322K, also identified as category II
sites that have evolved uniquely to a particular subtype when
sites in subtype K, are sites directly involved in or adjacent to
compared to the other subtypes. The branch site results were
sites directly involved in the switch from the CXCR4 to the
significant for the branches leading to subtypes A (P ϭ 0.02),
FIG. 2. Positions of category I sites (identified by ͉ for subtype F) and category II sites (identified by ∧ and ء for subtypes A and K, respectively)
across the gp120-coding sequence. Secondary protein structures are marked above their coding sequences. Site positions are described using theHXB2 reference sequence.
HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
FIG. 3. Positions of category I sites (identified by ϳ for subtype C) and category II sites (identified by ∧ and ء for subtypes A and K,
respectively) across the gp41-coding sequence. Secondary protein structures are marked above their coding sequences. Site positions are describedusing the HXB2 reference sequence. gp41 structural amino acids. Within gp41, many of the iden-
amino acid at that site in the radiation of the subtype. These
tified category I and category II sites are located in the C-
amino acid changes were radical, with large physiochemical
terminal transmembrane region for which, as yet, there is no
distances between them when compared using the Grantham
three-dimensional structure. Only one category I site was iden-
indices (10). For example, within subtype C, position 665 in the
tified (665K, subtype C) within the known gp41 ectodomain
gp41 flexible linker region contains a serine, while a lysine is
(Fig. 3) and was located in the crucial flexible linker region that
present in all other subtypes. Previous studies (33, 34) have
connects the ectodomain to the transmembrane region. Within
shown that functional shifts in a protein are often associated
category II, only one subtype A amino acid site (641L) and six
with amino acids that exhibit evidence of positive selection.
subtype K amino acid sites for were identified within the
Therefore, it is possible that these positively selected amino
acid changes, observed here in HIV-1 subtypes, may also haveinduced functional change. However, further analysis of the
DISCUSSION
effects on viral fitness and structure by the observed replace-ments is needed.
We have conducted an analysis of the intersubtype evolution
There was little correlation between category I sites and
of HIV-1 group M subtypes that identify groups of sites that
amino acid sites identified in other studies (4, 27–29) as hav-
have been subject to different selective constraints in the lin-
ing undergone strong selective pressures. Previous intersub-
eages leading to each subtype. The use of evolutionary models
type studies (4, 28) examined group M subtypes independently,
that incorporate different rates over different lineages allowed
looking at selective pressures within each subtype. In this study,
for the detection of sites undergoing evolutionary constraints
we have examined the selective pressures of the branches lead-
unique to a lineage within the data that would not have beendetected in any other method of analysis. Two categories of
ing to each subtype compared to those of all other lineages.
sites were observed: first, sites that have experienced positive
Sites we have identified as undergoing positive selection in one
selection in a particular subtype when the same site has expe-
subtype compared to all other subtypes may seem to be un-
rienced purifying selection in all other subtypes (category I);
dergoing purifying selection when only the subtype itself is ex-
and second, sites that have experienced purifying selection in
amined due to the conserved nature of these sites within a
one subtype while other subtypes have experienced positive
selection at that site (category II). Sites with a high probability
Category II amino acid sites. Both subtype A and subtype K
of being in category I were identified within subtypes C, F1,
contain amino acid sites that have been under pressure to
and G, while sites with a high probability of being in category
retain their current state, while these sites are under pressure
II were identified within subtypes A and K.
to change in all other subtypes. While a small number (six) of
Category I amino acid sites. Upon sequence examination,
such sites was identified for subtype A, a much larger number
category I sites are generally composed of amino acids that are
(45) was identified for subtype K. This finding may indicate a
conserved in the selected subtype but different in all other
substantial difference between the selective pressures govern-
subtypes. This indicates positive selection for this particular
ing the evolution of subtype K and those of all other subtypes.
Available molecular evidence indicates that subtype K has
REFERENCES
existed for as long as the other subtypes (24), but its observed
1. Abebe, A., C. L. Kuiken, J. Goudsmit, M. Valk, T. Messele, T. Sahlu, H.
worldwide prevalence is low. Only two subtype K full-genome
Yeneneh, A. Fontanet, F. De Wolf, and T. F. Rinke De Wit. 1997. HIV type 1 subtype C in Addis Ababa, Ethiopia. AIDS Res. Hum. Retrovir. 13:
sequences were available for use in this analysis, and this may
have had some effect on the analysis. The genetic distance be-
2. Abebe, A., G. Pollakis, A. L. Fontanet, B. Fisseha, B. Tegbaru, A. Kliphuis,
tween the two subtype K sequences was large (9%), while that
G. Tesfaye, H. Negassa, M. Cornelissen, J. Goudsmit, and T. F. Rinke de Wit. 2000. Identification of a genetic subcluster of HIV type 1 subtype C (CЈ)
of other subtypes used in this study, which also had only two
widespread in Ethiopia. AIDS Res. Hum. Retrovir. 16:1909–1914.
representative sequences (and yet did not yield a significant
3. Caffrey, M., M. Cai, J. Kaufman, S. J. Stahl, P. T. Wingfield, D. G. Covell,
branch site model LRT), was smaller; for example, there was
A. M. Gronenborn, and G. M. Clore. 1998. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 17:4572–4584.
only a 2% genetic distance between the two A2 representa-
4. Choisy, M., C. H. Woelk, J. F. Guegan, and D. L. Robertson. 2004. Com-
tive sequences. When subtype K env gene fragments available
parative study of adaptive molecular evolution in different human immuno-
through the Los Alamos National Laboratory HIV sequence
deficiency virus groups and subtypes. J. Virol. 78:1962–1970.
5. Descamps, D., C. Apetrei, G. Collin, F. Damond, F. Simon, and F. Brun-
database (http://hiv-web.lanl.gov) were used in a similar anal-
Vezinet. 1998. Naturally occurring decreased susceptibility of HIV-1 subtype
ysis, the branch site model was not significant (data not shown).
G to protease inhibitors. AIDS 12:1109–1111.
This lack of lineage-specific model significance is most likely
6. Drummond, A., O. G. Pybus, and A. Rambaut. 2003. Inference of viral
evolutionary rates from molecular sequences. Adv. Parasitol. 54:331–358.
due to the much shorter sequence data used (150 codons) and
7. Esparza, J., and N. Bhamarapravati. 2000. Accelerating the development
the fact that the fragments covered the V3-V5 region, where
and future availability of HIV-1 vaccines: why, when, where, and how?
only 20% of category II sites were observed in the full gene.
Lancet 355:2061–2066.
8. Fares, M. A. 2004. SWAPSC: sliding window analysis procedure to detect
A strong correlation was observed between sites identified as
selective constraints. Bioinformatics 20:2867–2868.
category II amino acids in this study and sites observed as
9. Gao, F., E. Bailes, D. L. Robertson, Y. Chen, C. M. Rodenburg, S. F.
undergoing strong evolutionary constraints in other studies (4,
Michael, L. B. Cummins, L. O. Arthur, M. Peeters, G. M. Shaw, P. M. Sharp, and B. H. Hahn. 1999. Origin of HIV-1 in the chimpanzee Pan troglodytes
27–29). For example, 50% of the subtype K category II sites in
troglodytes. Nature 397:436–441.
the gp120 three-dimensional structure were determined to be
10. Grantham, R. 1974. Amino acid difference formula to help explain protein
undergoing strong selective pressures in at least one of the
evolution. Science 185:862–864.
11. Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A.
other studies examined (4, 27–29) (Table 2). This is not sur-
Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in
prising, as category II sites indicate positive selection in all
complex with the CD4 receptor and a neutralizing human antibody. Nature
subtypes other than the one of interest. The other studies also
393:648–659.
12. Maddison, W. P., and D. R. Maddison. 1992. MacClade, version 4.5. Sinauer
identified positive selection at many of the same sites within
the same subtypes as those identified in this study.
13. Moore, J. P., F. E. McCutchan, S. W. Poon, J. Mascola, J. Liu, Y. Cao, and Functionality of category I and category II sites. Amino acid D. D. Ho. 1994. Exploration of antigenic variation in gp120 from clades A through F of human immunodeficiency virus type 1 by using monoclonal
sites critically important in gp120 and gp41 function such as
antibodies. J. Virol. 68:8350–8364.
receptor binding are for the most part undergoing similar evo-
14. Novitsky, V. A., M. A. Montano, M. F. McLane, B. Renjifo, F. Vannberg,
lutionary mechanisms within the subtypes as determined in this
B. T. Foley, T. P. Ndung’u, M. Rahman, M. J. Makhema, R. Marlink, and M. Essex. 1999. Molecular cloning and phylogenetic analysis of human immu-
study. The intersubtype evolutionary differences have been ob-
nodeficiency virus type 1 subtype C: a set of 23 full-length clones from
served mostly at amino acid sites involved in structure. A num-
Botswana. J. Virol. 73:4427–4432.
15. Peeters, M., and P. M. Sharp. 2000. Genetic diversity of HIV-1: the moving
ber of exceptions to this observation included sites involved in
target. AIDS 14(Suppl. 3):S129–S140.
direct CD4 binding, sites implicated in CXCR4-to-CCR5 co-
16. Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model of
receptor switch, and a putative glycosylation site (category II
DNA substitution. Bioinformatics 14:817–818.
17. Renjifo, B., B. Chaplin, D. Mwakagile, P. Shah, F. Vannberg, G. Msamanga,
sites determined in subtype K). Also, within the flexible linker
D. Hunter, W. Fawzi, and M. Essex. 1998. Epidemic expansion of HIV type
of the gp41 (important for tethering gp41 to the transmem-
1 subtype C and recombinant genotypes in Tanzania. AIDS Res. Hum.
brane segment, coreceptor binding, and host cell entry), amino
Retrovir. 14:635–638.
18. Robertson, D. L., J. P. Anderson, J. A. Bradac, J. K. Carr, B. Foley, R. K.
acids in category I (665K, subtype C) and category II (668S
Funkhouser, F. Gao, B. H. Hahn, M. L. Kalish, C. Kuiken, G. H. Learn, T.
and 671N, subtype K) were observed. Three category II sites
Leitner, F. McCutchan, S. Osmanov, M. Peeters, D. Pieniazek, M. Salminen,
(295N, 297T, and 334S) identified within subtype K correspond
P. M. Sharp, S. Wolinsky, and B. Korber. 2000. HIV-1 nomenclature pro- posal. Science 288:55–56.
to one cluster of the 2G12 epitope, an antibody that has been
19. Rodenburg, C. M., Y. Li, S. A. Trask, Y. Chen, J. Decker, D. L. Robertson,
implicated in neutralization of multiple subtypes (25). M. L. Kalish, G. M. Shaw, S. Allen, B. H. Hahn, and F. Gao. 2001. Near
To our knowledge, this is the first study to examine the
full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents. AIDS Res. Hum. Retrovir. 17:161–168.
selective pressures that governed the evolution of the subtypes
20. Seo, T. K., J. L. Thorne, M. Hasegawa, and H. Kishino. 2002. Estimation of
of HIV-1 group M. We have identified categories of sites that
effective population size of HIV-1 within a host: a pseudomaximum-likeli-
have evolved under unique selective pressures for particular
hood approach. Genetics 160:1283–1293.
21. Shankarappa, R., R. Chatterjee, G. H. Learn, D. Neogi, M. Ding, P. Roy, A.
subtypes that may cause subtype-specific genetic characteris-
Ghosh, L. Kingsley, L. Harrison, J. I. Mullins, and P. Gupta. 2001. Human
tics. The presence of such sites indicates heterogeneity of se-
immunodeficiency virus type 1 env sequences from Calcutta in eastern India:
lective pressures within HIV evolution, and this fact should be
identification of features that distinguish subtype C sequences in India from other subtype C sequences. J. Virol. 75:10479–10487.
taken into account in any future HIV vaccine or treatment
22. Soares, M. A., T. De Oliveira, R. M. Brindeiro, R. S. Diaz, E. C. Sabino, L. Brigido, I. L. Pires, M. G. Morgado, M. C. Dantas, D. Barreira, P. R. Teixeira, S. Cassol, and A. Tanuri. 2003. A specific subtype C of human immunodeficiency virus type 1 circulates in Brazil. AIDS 17:11–21.
23. Swofford, D. L. 1998. PAUPء. Phylogenetic analysis using parsimony (ءand ACKNOWLEDGMENT
other methods), version 4.0. Sinauer Associates, Sunderland, Mass.
24. Triques, K., A. Bourgeois, N. Vidal, E. Mpoudi-Ngole, C. Mulanga-Kabeya,
Many thanks to Mario Fares for assistance in the use of SWAPSC
N. Nzilambi, N. Torimiro, E. Saman, E. Delaporte, and M. Peeters. 2000.
for synonymous substitution saturation detection.
Near-full-length genome sequencing of divergent African HIV type 1 sub-
HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
type F viruses leads to the identification of a new HIV type 1 subtype
in the human immunodeficiency virus type 1 genome. J. Mol. Evol. 57:
designated K. AIDS Res. Hum. Retrovir. 16:139–151.
25. Trkola, A., M. Purtscher, T. Muster, C. Ballaun, A. Buchacher, N. Sullivan,
29. Yang, Z. 2001. Maximum likelihood analysis of adaptive evolution in HIV-1 K. Srinivasan, J. Sodroski, J. P. Moore, and H. Katinger. 1996. Human
gp120 env gene. Pac. Symp. Biocomput. 2001:226–237.
monoclonal antibody 2G12 defines a distinctive neutralization epitope on the
30. Yang, Z. 1997. PAML: a program package for phylogenetic analysis by
gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:
maximum likelihood. Comput. Appl. Biosci. 13:555–556.
31. Yang, Z., and J. P. Bielawski. 2000. Statistical methods for detecting molec-
ular adaptation. Trends Ecol. Evol. 15:496–503.
26. Van Harmelen, J. H., E. Van der Ryst, A. S. Loubser, D. York, S. Madurai,
32. Yang, Z., and R. Nielsen. 2002. Codon-substitution models for detecting S. Lyons, R. Wood, and C. Williamson. 1999. A predominantly HIV type 1
molecular adaptation at individual sites along specific lineages. Mol. Biol.
subtype C-restricted epidemic in South African urban populations. AIDS
Evol. 19:908–917.
Res. Hum. Retrovir. 15:395–398.
33. Yang, Z., R. Nielsen, N. Goldman, and A. M. Pedersen. 2000. Codon-sub-
27. Yamaguchi-Kabata, Y., and T. Gojobori. 2000. Reevaluation of amino acid
stitution models for heterogeneous selection pressure at amino acid sites.
variability of the human immunodeficiency virus type 1 gp120 envelope
Genetics 155:431–449.
glycoprotein and prediction of new discontinuous epitopes. J. Virol. 74:
34. Zhang, J., Y. P. Zhang, and H. F. Rosenberg. 2002. Adaptive evolution of
a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat.
28. Yang, W., J. P. Bielawski, and Z. Yang. 2003. Widespread adaptive evolution
Genet. 30:411–415.
Epilepsia, ** (*):1–14, 2008 doi: 10.1111/j.1528-1167.2008.01765.x SPECIAL REPORT Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group ∗Eric H. Kossoff, † Beth A. Zupec-Kania, ‡ Per E. Amark, § Karen R. Ballaban-Gil, ¶ A. G. Christina Bergqvist, #Robyn Blackford, ∗∗Jeffrey R
 HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
TABLE 1. Representative sequences of selected subtypes
contrasted with all other subtypes using the branch site-specific models. The
branches leading to subtypes A, B, C, D, F, G, H, J, and K were labeled in a sep-arate analysis, as were the branches leading to the A1, A2, F1, and F2 lineages.
HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
TABLE 1. Representative sequences of selected subtypes
contrasted with all other subtypes using the branch site-specific models. The
branches leading to subtypes A, B, C, D, F, G, H, J, and K were labeled in a sep-arate analysis, as were the branches leading to the A1, A2, F1, and F2 lineages.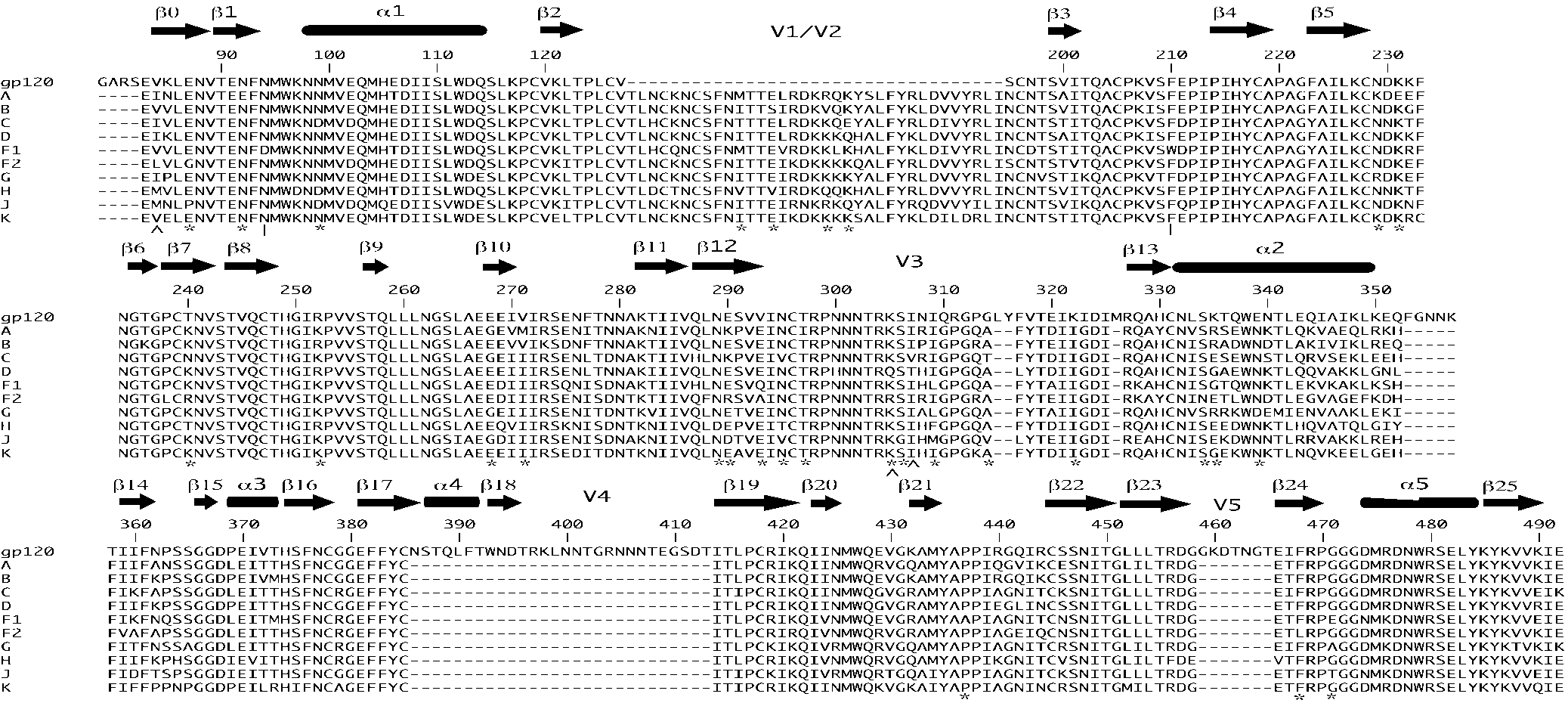 TABLE 2. Selected sites for each subtypea
C (P ϭ 0.01), F1 (P ϭ 0.005), G (P ϭ 0.02), and K (P ϭ 0.02),suggesting that these subtypes contain a category of sites that
have evolved differently from the other subtypes. From the
branch-specific model, the subtypes’ results fell into two cate-gories. In subtypes C, F1, and G, a proportion of their sites
were observed to have undergone positive selection, whereas
all other subtypes had undergone purifying selection at thatsite (described herein as category I sites). In subtypes A and K,
a proportion of sites was observed to have undergone purifying
selection with positive selection having occurred in the other
subtypes at those sites (described herein as category II sites).
TABLE 2. Selected sites for each subtypea
C (P ϭ 0.01), F1 (P ϭ 0.005), G (P ϭ 0.02), and K (P ϭ 0.02),suggesting that these subtypes contain a category of sites that
have evolved differently from the other subtypes. From the
branch-specific model, the subtypes’ results fell into two cate-gories. In subtypes C, F1, and G, a proportion of their sites
were observed to have undergone positive selection, whereas
all other subtypes had undergone purifying selection at thatsite (described herein as category I sites). In subtypes A and K,
a proportion of sites was observed to have undergone purifying
selection with positive selection having occurred in the other
subtypes at those sites (described herein as category II sites). HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
FIG. 3. Positions of category I sites (identified by ϳ for subtype C) and category II sites (identified by ∧ and ء for subtypes A and K,
respectively) across the gp41-coding sequence. Secondary protein structures are marked above their coding sequences. Site positions are describedusing the HXB2 reference sequence.
HETEROGENEOUS EVOLUTION BETWEEN HIV-1 GROUP M SUBTYPES
FIG. 3. Positions of category I sites (identified by ϳ for subtype C) and category II sites (identified by ∧ and ء for subtypes A and K,
respectively) across the gp41-coding sequence. Secondary protein structures are marked above their coding sequences. Site positions are describedusing the HXB2 reference sequence.